
The outcome of a viral infection is influenced by the balance between host defense mechanisms and virus modulation of cellular pathways. By making use of finely tuned protein interactions, viruses succeed in sabotaging intricate host processes and turning cellular systems to their own use. Although virus-host interactions have been extensively studied, our knowledge of the detailed virus-host protein interactions at the heart of these mechanisms remains quite limited. Our laboratory is interested in approaching the dynamic viral infection by tracking the localization and interactions of viral proteins in host systems as a function of infection time.
The work in our laboratory seeks to push the boundaries of current proteomic techniques by devising new tools for exploring protein interactions and placing them in defined biological contexts. We have developed a series of multidisciplinary strategies to be used in the context of viral infection, including identifying protein-protein and protein-nucleic acid interactions, quantifying subtle changes in protein interactions, defining distinct protein complexes, measuring the relative stability and the specificity of interactions, and defining the function of post-translational modifications. The proteomic approaches employed in our laboratory add a powerful new dimension to the already considerable reach of molecular biology in querying nature. Employing new ways to look at long-standing questions is one of the strongest leitmotifs in science, giving us new opportunities to appreciate, as Darwin saw it, the “grandeur in this view of life.”
We have successfully implemented our multidisciplinary methodologies to uncover the viral-host protein interactomes of a broad range of viruses, such as HCMV, HSV-1, KSHV, Influenza A, Coronavirus OC43, and HIV-1. Our findings have led to new hypotheses as to how viruses manipulate host cellular processes, encompassing everything from chromatin remodeling to reorganization of the subcellular landscape. The richness of novel identified interactions reminds us that we are still in virtually uncharted territory in understanding the complexity of the virus-host relationship.
Nuclear sensing of viral DNA en route to innate immune signaling
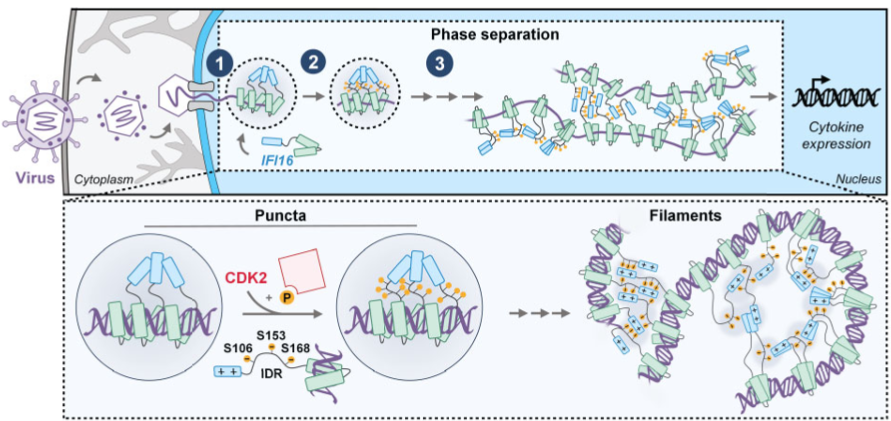
The ability of mammalian cells to recognize foreign DNA is an essential step for the onset of immune responses against viruses. This recognition is accomplished by proteins called sensors, which bind viral DNA and induce cytokine secretion to alert neighboring cells and inhibit the spread of infection. Given the conceptual challenge of distinguishing two similar molecules (viral and cellular DNA), the sensing of viral DNA was thought to occur only in subcellular compartments typically devoid of host DNA (e.g., cytoplasm). However, this long-standing belief failed to fully explain how the cell detects nuclear-replicating DNA viruses. Our lab’s characterization of the first identified nuclear DNA sensor, IFI16, has helped to establish the novel concept of nuclear sensing. We demonstrated that nuclear IFI16 senses nuclear-replicating herpesviruses, including HCMV and HSV-1. We also defined the viral immune evasion mechanisms, demonstrating that HCMV inhibits IFI16 oligomerization and the propagation of immune signals, while HSV-1 targets IFI16 for degradation. We further described how IFI16 oligomerization is paired with a liquid-liquid phase separation mechanism dependent on the IFI16 IDR, which is crucial for initiation of cytokine responses. The discovery that sensing can occur in the nucleus opens a new direction for research in immunity that will ultimately increase our understanding of how balanced immune responses work to maintain a healthy system and how their mis-regulation leads to immune disorders, cancers, and virus-induced mortality.
Interplay between pattern recognition receptors and DNA-damage response signaling
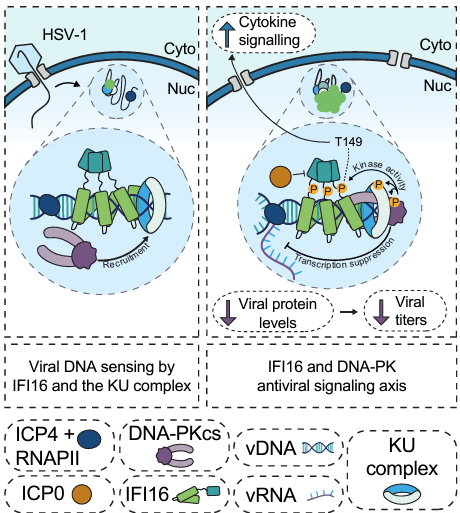
Pattern-recognition receptors, such as IFI16, do not act alone to transduce antiviral immune signaling. We have shown that IFI16 interacts with another DNA sensor, cGAS, to bolster immune signaling through the STING-TBK1-IRF3 axis. Further, IFI16 coordinates inflammatory responses to DNA damage outside of the context of infection by interaction with p53. We dissected how IFI16 coordinates with master DNA-damage response (DDR) kinases ATM and DNA-PK to initiate cytokine responses during DNA damage, as well as how IFI16 contributes to the DNA-PK-driven DDR landscape during herpesvirus infection. Continued investigation of the interface between IFI16 and DDR signaling may unravel the architecture of the IFI16 immune signaling axis, with likely implications for autoinflammatory diseases and cancer.
Virus-induced organelle remodeling
Organelle remodeling is an essential component of all human virus infections. Modulation of cellular organelles allows virion entry and trafficking, suppresses immune signaling, provides structural support of virus replication compartments, and rewires cellular metabolism for optimal virion production. At the same time, host cells utilize organelles and organelle-resident proteins to detect and combat pathogens. How organelle remodeling contributes to viral pathogenesis, as well as host intrinsic and innate responses to infection, is a longstanding interest of our group. In the process, we have uncovered regulatory mechanisms conserved in other disease states and normal cellular physiology.
Organelle structure
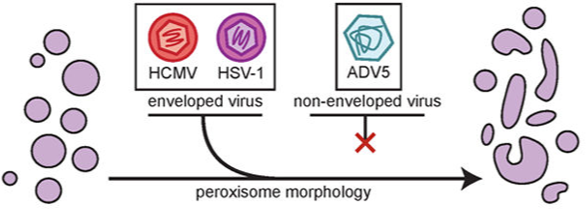
HCMV restructures most cellular organelles throughout its replication cycle. We found that HCMV infection polarizes peroxisomes into large and small sub-populations. HCMV virion production relies on the increased synthesis of specific lipids for the viral envelope, including peroxisome-derived plasmalogens, and we found that the disruption in normal peroxisome morphology underlies this change. Additionally, mitochondria are extensively fragmented during infection. Fragmentation is typically a hallmark of sick mitochondria, but surprisingly, HCMV infection is characterized by an increase in oxidative phosphorylation in the mitochondria. Through prior subcellular proteomic studies, we found that the viral protein pUL13 traffics to the mitochondria during infection. We discovered that pUL13 supports increased oxidative phosphorylation through alteration in cristae ultrastructure, the first viral protein ever found to employ this mechanism.
Organelle-organelle interactions
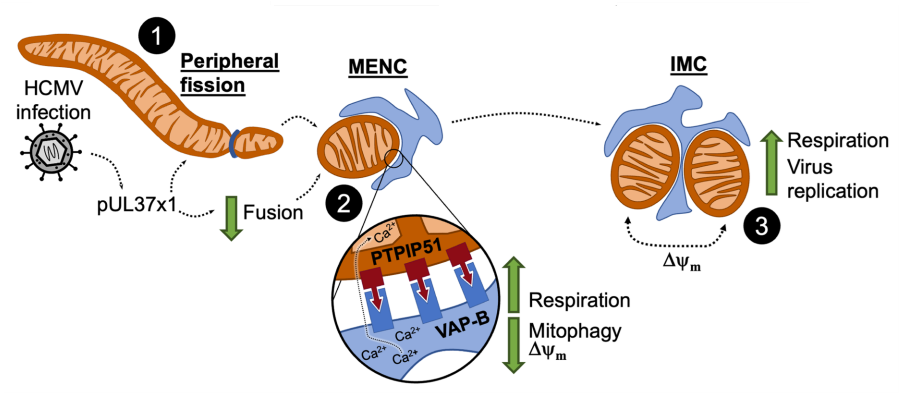
Beyond deformation of isolated organelles, viruses alter organelle-organelle communication to modulate organelle structure and function. This communication is enabled by membrane contact site (MCS) proteins that facilitate tethering of organelles and transfer of biomolecules. We found that viruses differentially regulate MCS protein abundance to remodel the organellar landscape. Following on the quandary of increased mitochondrial bioenergetics from fragmented mitochondria, we revealed a novel contact site called a mitochondria-ER encapsulation (MENC), which becomes the predominant mitochondria-ER contact by the late stages of HCMV infection. By preventing mitophagy of fragmented mitochondria and supporting calcium flux from the ER, MENCs support high levels of respiration. We found that MENCs additionally support respiration in certain cancers, in which MENC formation correlates with severe cancer malignancy.
Intercellular communication in the virus microenvironment
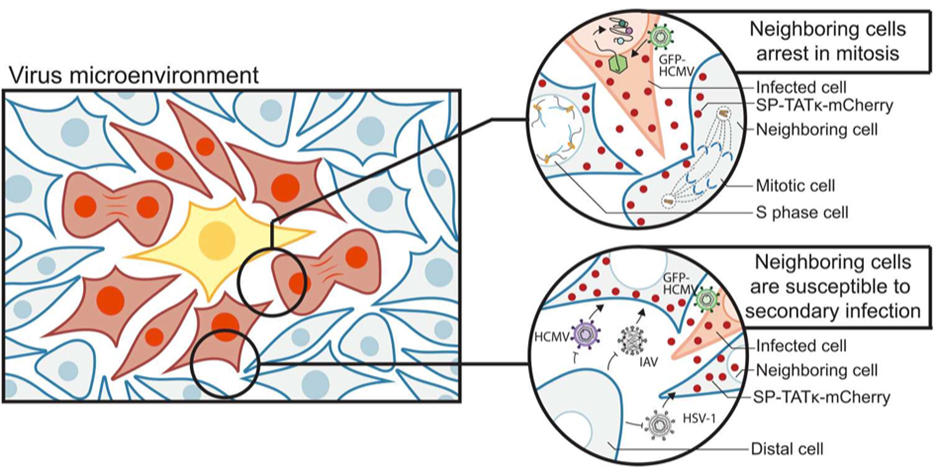
Beyond virus manipulation of host intracellular processes, viruses control intercellular communication. In order to interrogate how viruses influence nearby uninfected cells, we adapted a technology from the tumor microenvironment which allows us to isolate cells proximal to the primary infection center. Through proteomic analysis of infected cells, proximal cells, and cells distal to the infected cell, we elucidated how virus-infected cells modulate their cellular microenvironment. We found that infection leads to cell cycle dysregulation and a dampened immune response in nearby cells, paired with an increased susceptibility of those nearby cells to subsequent infections by HCMV or co-infection with other viruses. This is induced in large part by exosomes trafficking viral proteins, mRNAs, and noncoding RNAs to nearby cells. We are excited to learn about the importance of these mechanisms in supporting spread of viruses, as well as how this relates to susceptibility to co-infection and perhaps the oncogenic or oncomodulatory effects of many viruses.
Spatiotemporal regulation of virus-host interactions
Protein interactions, of a stable or transient nature, underlie the majority of cellular processes. The composition and organization of protein complexes across space and time dictates their potential functions. Accordingly, eukaryotic cells spatially organize their proteomes to partition biological functions between the cytosol and specialized organelles, both membrane-bound and liquid-liquid phase separated. Viruses have evolved to overcome the challenge of replicating within this spatially-defined cellular landscape, and many aspects of their infection cycles are regulated or organized at the subcellular level. We seek to understand how spatial and temporal boundaries dictate virus-host protein interactions, and, in turn, how these interactions hinder or promote the viral infection cycle.
Protein-protein interactions
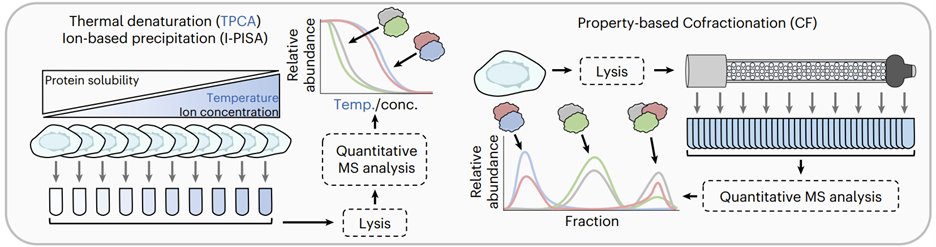
We developed an approach to define dynamic macromolecular interactions via a single fluorescent tag that allows us to both visualize proteins in live cells and capture their interacting partners with rapid, high-affinity immunopurifications. We found that this methodology opens new doors for almost every protein complex that we have approached, leading to novel insights into cellular processes as well as new questions. We have additionally been pioneering the use of global protein-protein interaction approaches, such as thermal proximity coaggregation assay (TPCA), to acquire a systems-level understanding of protein interaction dynamics during viral infections. We have employed machine learning pipelines to increase the scope and fidelity of these predictions, utilizing it to predict de novo protein-protein interactions that underlie viral replication mechanisms during HSV-1, HCMV, and KSHV infections.
Subcellular localization
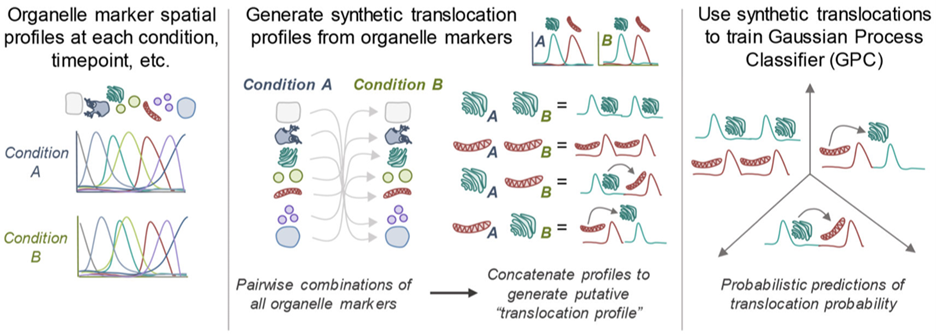
Viruses are known to alter protein subcellular localization to support viral replication and evade host immunity. We have utilized density-gradient fractionation during infection to globally profile proteome re-organization. Through machine-learning approaches, we predicted virus-induced shuttling events between organelles. Altogether, this work revealed that HCMV infection causes a striking global reorganization of the host proteome to regulate organelle composition and function, generating specialized infection-induced compartments. In addition, we were able to demonstrate the spatiotemporal dynamics of previously uncharacterized viral proteins, providing novel insights into their functions and importance for viral replication. This work highlighted the importance of subcellular organization during infection time and has prompted many ongoing follow-up studies into the dynamic, localization-dependent functions of both host and viral proteins. Continued investigation of infection-induced subcellular remodeling has also challenged us to develop new imaging and multidimensional analysis techniques to parallel the proteomic investigations of exchanges between subcellular compartments.
Post-translational modifications
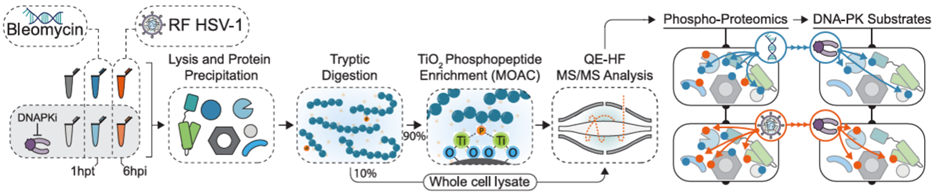
Through rapid toggling of protein functions with post-translational modifications (PTMs), viruses fine-tune the cellular proteome to facilitate viral replication and disable host immune responses. We have explored PTM dynamics during HCMV and HSV-1 infections, thus far focusing on acetylation and phosphorylation. We uncovered that lamin acetylation prevents egress of capsids from the infected cell nucleus. Further work found that this acetylation site naturally fluctuates with the cell cycle, slowing the rate of nonhomologous end-joining to stall G1-to-S cell cycle progression. Then, through a comparative analysis of the phospho-proteome during HSV-1 infection or upon induced DNA double-strand breaks, we have additionally explored the context-dependence of the cellular cytokine response to DNA damage. By surveying the PTM landscape during infection, we have uncovered pro- and anti-viral mechanisms with origins in general cell biology.

